




|
关于目标区域测序 目标区域测序(Targeted Sequencing):是目标区域测序是指针对感兴趣的目标区域富集后进行大规模测序。研究者可以针对自己感兴趣的染色体区域或者大量的候选基因区域进行数百个甚至上千个样品的序列测定。 目标区域测序优势 针对性强:比起全基因组水平的研究,目标区域测序更具有针对性,可以依赖大量的前期研究成果,获得候选染色体区域或者基于生物通路的大量候选基因。 费用低:目标区域测序区域较小,可对数百个样品进行快速测序,大大降低了研究成本。 信息量大:比起目标区域或者候选基因单倍型标签SNP分型的研究策略,目标区域测序可以完整覆盖整个基因区域,不仅可以获得高频SNP的分型数据,而且还可以发现低频的和个体特有的变异。 效率高:比起使用Sanger法的候选基因测序方法,基于二代测序技术的目标区域测序更加快速、高效! 高精度:目标区域的高测序深度保证了更准确的测序结果,例如目标区域测序的测序深度可以达到200×。 目标区域测序在动植物研究中的应用 1.有些物种是异源四倍体物种 ,对于这种异源四倍体物种其一个基因特定位点最多有四种不同的等位基因,因此要准确区别不同的等位基因和准确确定等位基因的拷贝数在测序时相对于二倍体就需要更高的测序深度,其测序深度至少要达到48×,此时目的目的区域测序就显示出其无可比拟的优势。通过对这些异源多倍体物种的目标区域进行富集捕获测序,数据可用于群体结构以及GWAS-QTL分析。 相应研究案例: 对83株四倍体栽培土豆和1株参照二倍体土豆,平均分布在基因组上807个基因,共2.1M的区间进行富集,测序后获得平均覆盖深度为63×,共12.4G的高质量数据,发现129,156个可靠的序列变异(在外显子1 SNP/24 bp ,在内含子1 SNP/15 bp),这些变异含有大量罕见变异(61%的变异MAF小于 0.05),经KASP技术验证有99%的一致性。利用发现的变异对土豆植株成熟期和块茎肉色相关的QTL进行GWAS分析,定位到了之前的已知QTL位点。主成分分析发现栽培土豆可以明显聚类为五组。 
图1. DNA序列变异与土豆的两种性状:(A)植株成熟期和(B)块茎颜色相关性p值的Manhattan图
参考文献:A Next-Generation Sequencing Method for Genotyping-by-Sequencing of Highly Heterozygous Autotetraploid Potato. Jan G. A. M. L. Uitdewilligen et al. 2013, PloS ONE.2.针对某些物种间的保守区域进行目标区域测序,利用测序数据进行物种分类和系统进化分析,这种策略类似于利用16S rDNA/18S rDNA/ITS扩增子测序进行微生物群落多样性分析或利用DNA条形码技术进行品种资源鉴定和系统进化分析。 相应研究案例: 不同鸟类的演化仍然有争议,利用杂交富集的定向测序技术,对198种现存鸟类(代表所有鸟类谱系和两个鳄鱼外群)的394个有足够变异度的保守位点进行目标区域测序(Agilent定制液相芯片富集),然后基于测序数据使用贝叶斯法和最大似然分析法建立所有鸟类谱系的系统进化树。产生259个高质量测序核位点(平均组装长度为1523bp)共7.8 × 107 个碱基的数据量,基于这些数据使用贝叶斯法和最大似然分析法建立所有鸟类谱系的进化树,5个主要分支形成新鸟纲的连续姐妹类群:(1)包括夜鹰,雨燕和蜂鸟;(2)包括杜鹃,大鸨,鸽子,蕉鹃和沙鸡;(3) 鹤及其亲属;(4)水鸟类群,包括潜水类、涉水类、岸滩类;(5)麝雉类(图1)。 
图1. 鸟类的系统发育树
参考文献:A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Richard O. Prum et al. 2015, Nature.3.为了保护种质资源,开发可靠的并且高度可变的遗传标记是必不可少的,对于一些基因组复杂但是遗传标记比较缺少的物种,过去常使用细胞器基因组进行相关系统进化研究,但是这种方法具有效率低和单亲遗传等缺点,因此目前高通量测序非常适合这种非模式物种的遗传标记开发研究,但是使用全基因组重测序对这类基因组复杂物种进行群体进化研究实际上是代价高昂的,而目标区域测序这种只针对某些可靠位点进行深度测序的技术实际上更符合成本效益。 相应研究案例: 根据白皮松转录组序列设计捕获探针,从而对48棵白皮松(每棵白皮松代表不同地理位置)上的7,849个不同基因进行目标区域捕获测序,从这48棵白皮松样本中共得到 390,910,265条 reads,所得到的数据提供了4452个基因的基因信息,共鉴定到12390个多态性位点(其中2163个变异位点的MAF > 0.1),然后通过这些位点揭示了杂合度和等位基因丰度的地理分布趋势, PCA分析结果与这48棵白皮松的实际地理分布是一致的,并且指出南部树木相对于其它区域的树木显示出最大的差异分化。 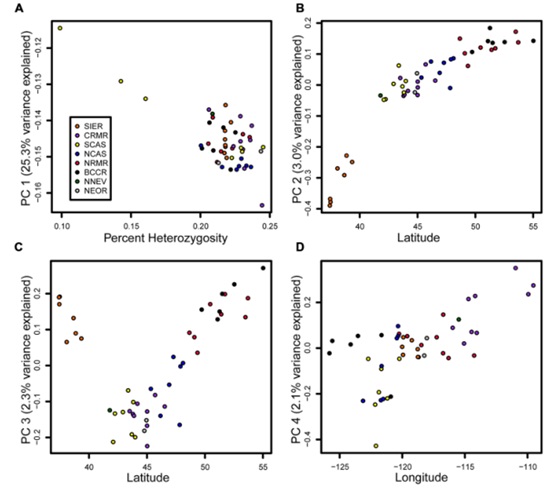
图1. 与杂合性,纬度和经度相关的主成分(PC)。样品的颜色按地理分布,(a)PC1与杂合度。(b)PC2与纬度。(C)PC3与纬度。(d)PC4与经度
参考文献:Targeted Capture Sequencing in Whitebark Pine Reveals Range-Wide Demographic and Adaptive Patterns Despite Challenges of a Large, Repetitive Genome.Syring JV et al. 2016,Front Plant Sci. 其它应用展望 4. 传统方法QTL性状粗定位锁定大致区域后,如果区域内没有足够多的分子标记或者没有合适的分子标记,可使用目标区域测序来开发分子标记从而进行下一步的精细定位,如果精细定位锁定区域大小在目标区域测序大小范围内可以使用目标区域测序直接锁定QTL关联位点或基因。 5. 混池分组分析法(BSA)已经广泛应用于动植物QTL定位的研究中,并且发现了QTL候选区域,因此后续可以对候选区间进行目标区域测序,获得区域内SNP/InDel的多态信息,联合性状进行关联或者连锁分析,从而锁定QTL关联位点/基因。 6. 全基因组关联分析(GWAS)己经广泛地应用于QTL性状的研究当中,并且定位出了大量显著的SNP位点,然而这些标记位点大部分为常见变异位点,因此可以通过对 GWAS 鉴定的区域进行目标区域测序,从而找到与QTL性状紧密相关的其它新的、稀有的和可能的功能变异。 目标区域测序在动植物研究中的应用总结 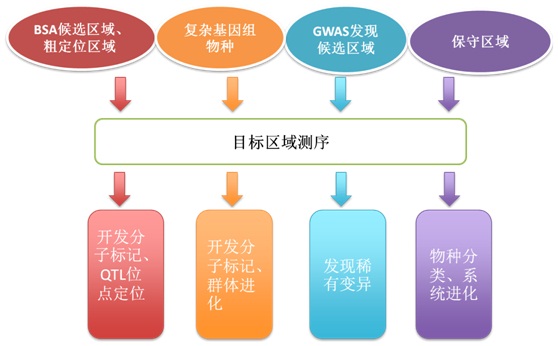 天昊生物目标区域测序整体解决方案 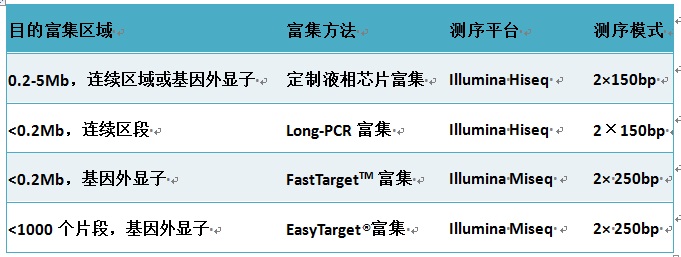 天昊生物目标区域测序特色
天昊目标区域测序技术发表高分文章
FastTargetTM项目实例: 项目简介:利用FastTargetTM富集技术对32个基因(约140K区域)350个样本进行测序。 数据量统计:共测序得到37M的reads,有效34M的reads,富集效率92%,平均每个片段覆盖173X。 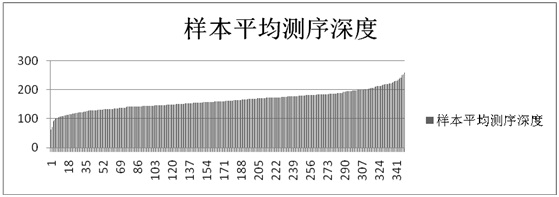 数据结果验证:采用SNaPshot对目标区域内12个高频SNP位点对所有样本进行了分析,观察基因型的一致性,除了少部分样本因为测序深度不够,其它基本上达到了100%的一致性。 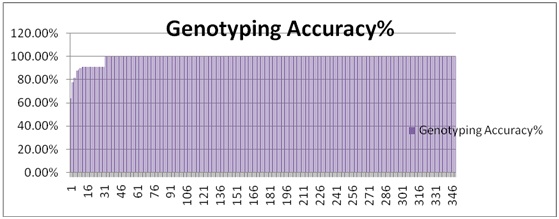 EasyTarget®项目实例 项目简介:采用EasyTarget®富集方法对 9个基因(约37K区域)133个样本进行测序。 数据量统计:总数据量为11M reads,有效reads为9.6M,富集效率87%,平均每个样本72K reads,平均每个片段覆盖486X,样本测序深度分布如下。  数据结果验证:全部通过一代Sanger测序验证,100%的准确性。 |


